
[ad_1]
Dans une étude récemment publiée dans la revue PNAS, Les chercheurs ont étudié comment l’élevage à grande échelle peut conduire à l’émergence et à la transmission de nouveaux agents pathogènes potentiellement zoonotiques. Ils ont combiné plusieurs sources de données issues de la datation moléculaire, de la génomique comparative et de la phylogéographie pour examiner comment l’élevage porcin a permis de nouveaux développements. Streptococcus suis Des lignées dont certaines sont capables de retombées zoonotiques. Leurs résultats montrent comment les agents pathogènes, notamment S. suiss’adapter pour exploiter des changements significatifs dans la taille de leur population hôte et comment cela, à son tour, peut contribuer indirectement à l’émergence de nouvelles souches hautement pathogènes, potentiellement pandémiques et zoonotiques, de microbes auparavant inoffensifs.
 Étude: L’émergence et la diversification d’un pathogène zoonotique à partir du microbiote de porcs d’élevage intensif. Crédit photo : Dusan Petkovic / Shutterstock
Étude: L’émergence et la diversification d’un pathogène zoonotique à partir du microbiote de porcs d’élevage intensif. Crédit photo : Dusan Petkovic / Shutterstock
L’élevage et ses effets sur les agents pathogènes
Au cours des derniers siècles, l’augmentation explosive de la population humaine et le besoin associé de bétail pour se nourrir et travailler pour soutenir l’industrie agricole ont conduit à une expansion mondiale de l’élevage. La boucle de rétroaction agricole des systèmes agricoles intensifs permet d’augmenter le cheptel, ce qui nécessite à son tour une production agricole accrue. Cela a eu pour conséquence que les populations de bétail dépassent désormais les populations humaines et sauvages totales.
Ces pratiques, combinées au transport sur de longues distances d’animaux reproducteurs, ont contribué à une faible diversité génétique et à une densité élevée du cheptel. Cela représente une recette idéale pour des épidémies d’agents pathogènes qui peuvent anéantir des millions de bétail sans capacité de résistance génétique, et dont le transport peut infecter non seulement d’autres populations de bétail, mais également des populations sauvages de la même espèce ou d’espèces similaires.
L’hypothèse selon laquelle ce cocktail d’événements favorise l’émergence de nouveaux agents pathogènes zoonotiques, résultant de la propagation de l’agent pathogène vers de nouveaux hôtes et de mutations dans un microbiote auparavant inoffensif auparavant associé aux animaux d’élevage, est préoccupante.
“Cette voie d’émergence d’agents pathogènes peut être particulièrement importante dans les systèmes agricoles intensifs, où de grandes populations et des densités de population élevées peuvent sélectionner des traits associés à la pathogénicité, tandis que la biosécurité réduit le risque d’entrée de nouveaux agents pathogènes dans la population.”
Streptococcus suis est un composant omniprésent du microbiote des voies respiratoires supérieures des porcs. L’élevage porcin intensif et auparavant inoffensif au XIXe siècleÈme et 20Ème Pendant des siècles, on a observé que le microbe s’adaptait à un mode de vie plus pathogène. En 1954, la bactérie a été associée à une maladie répandue chez les porcs et est aujourd’hui l’une des maladies les plus courantes chez les porcelets. De manière inquiétante, la bactérie s’est propagée de manière zoonotique à l’homme en raison de l’accumulation de mutations, provoquant des méningites, de l’arthrite, des endocardites et des septicémies. a entraîné une mort subite chez les humains et les porcs.
Suivez la première personne S. suisAprès avoir provoqué une mortalité associée en 1968, la bactérie a connu depuis des épidémies majeures en Chine et est l’une des principales causes de septicémie et de méningite chez les adultes dans toute l’Asie du Sud-Est.
« Les difficultés rencontrées pour identifier les facteurs de pathogénicité de S. suis ont été attribuées à la pathogenèse complexe et à la grande diversité génétique. “Peu d’études ont pris en compte les facteurs de virulence dans des souches autres que ST 1, responsable de la plupart des cas de maladie à S. suis chez le porc et chez l’homme dans le monde.”
À propos de l’étude
Dans la présente étude, les chercheurs ont examiné les liens entre l’élevage porcin intensif et l’émergence de nouveaux porcs. S. suis Les lignées et leur potentiel de retombées zoonotiques. Ils ont effectué une analyse génomique de population de plus de 3 000 échantillons bactériens obtenus à partir d’écouvillons d’amygdales et nasaux de porcs et de sangliers. En outre, ils ont collecté du sang infecté provenant de personnes et de porcs souffrant de la maladie à S. suis en Amérique du Nord, en Europe, en Asie et en Australie. Leur objectif était d’élucider les origines, la répartition géographique et le degré de diversification des lignées pathogènes des bactéries.
L’ensemble de données de l’étude comprenait 3 070 isolats génomiques de S. suis Échantillons dérivés de données précédemment publiées et collectés et séquencés dans le cadre de ce projet. Ceux-ci comprenaient 29 génomes de référence publiés et des isolats de collection provenant de 15 pays répartis sur les cinq continents mentionnés ci-dessus. L’échantillonnage a eu lieu entre 2014 et 2018. Les isolats ont été traités via le pipeline de séquençage du génome entier Illunima HiSeq 25000, qui a ensuite été utilisé pour construire une bibliothèque d’isolats génomiques. Les séquences brutes ont été vérifiées, nettoyées et utilisées pour générer des assemblages de novo pour les évaluations du polymorphisme.
L’étude d’Athey et al. Le pipeline décrit a été utilisé pour les analyses de sérotypage et de typage par séquençage. Les génomes générés ont ensuite été annotés pour identifier les gènes homologues et analyser les îlots génomiques associés à la pathogénicité. Le logiciel PopPunk a ensuite été utilisé pour identifier les génomes divergents et les classer en lignées. Les six lignées les plus courantes ainsi identifiées ont été testées pour détecter les signaux temporels en effectuant une régression des distances entre les racines et les pointes en fonction de l’année d’échantillonnage des isolats. Enfin, des reconstructions d’états ancestraux ont été utilisées pour déduire la répartition géographique des lignées identifiées.
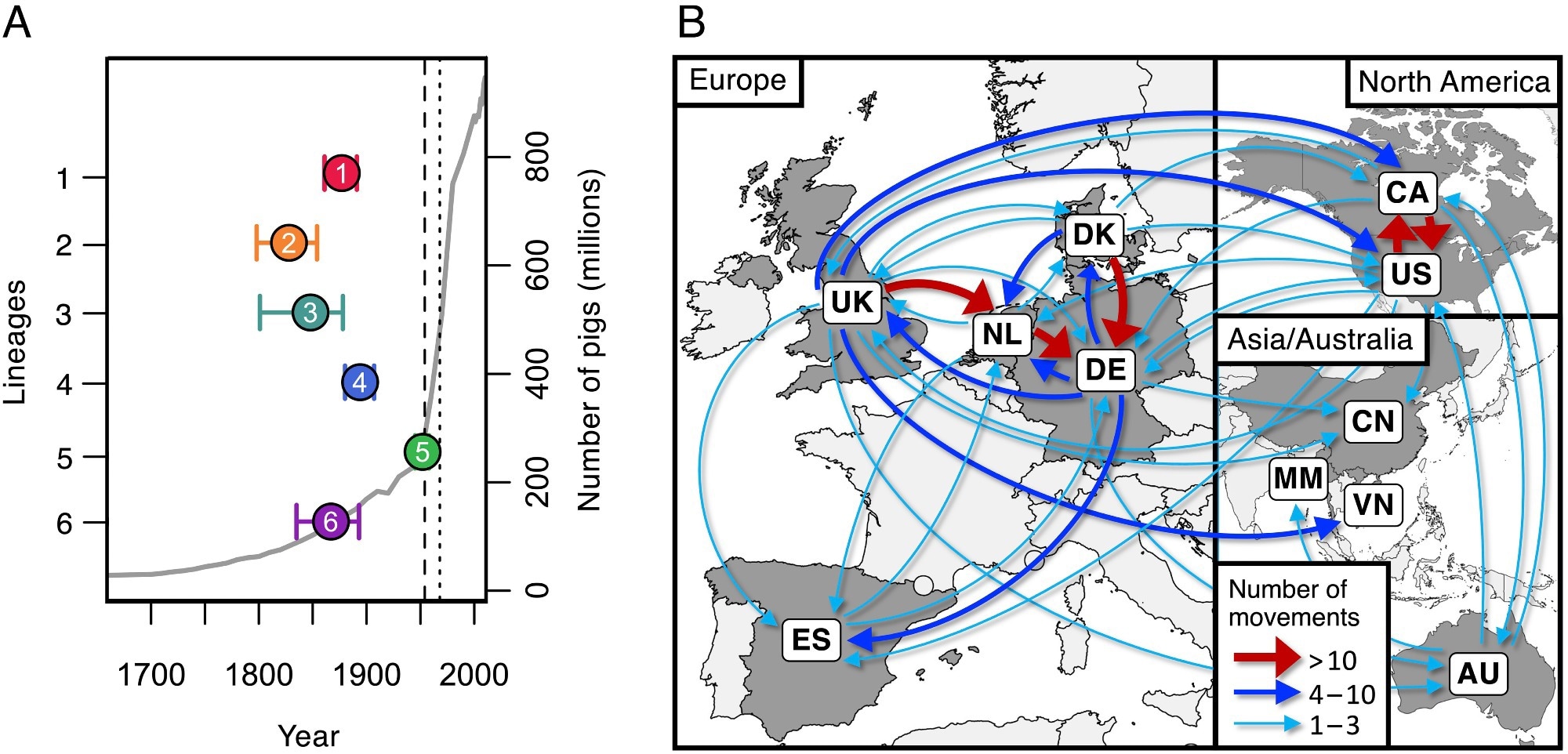 Dates d’origine et voies de transmission entre les pays pour les six lignées pathogènes les plus courantes. (A) Estimations des dates d’ancêtre commun les plus récentes des six lignées pathogènes les plus courantes (points colorés) par rapport à une estimation du nombre mondial de porcs (ligne grise). La ligne pointillée verticale indique la date du premier cas signalé de maladie à S. suis chez le porc (1954) et la ligne pointillée montre le premier cas signalé chez l’homme (1968). (B) Carte montrant les voies de transmission déduites de ces six lignées pathogènes entre les pays de notre collection. Les flèches représentent les itinéraires avec au moins un événement de transmission dérivé. Les itinéraires avec plus de dix événements de transmission dérivés sont affichés en rouge, les itinéraires avec plus de trois sont affichés en bleu et les itinéraires avec un à trois sont affichés en turquoise.
Dates d’origine et voies de transmission entre les pays pour les six lignées pathogènes les plus courantes. (A) Estimations des dates d’ancêtre commun les plus récentes des six lignées pathogènes les plus courantes (points colorés) par rapport à une estimation du nombre mondial de porcs (ligne grise). La ligne pointillée verticale indique la date du premier cas signalé de maladie à S. suis chez le porc (1954) et la ligne pointillée montre le premier cas signalé chez l’homme (1968). (B) Carte montrant les voies de transmission déduites de ces six lignées pathogènes entre les pays de notre collection. Les flèches représentent les itinéraires avec au moins un événement de transmission dérivé. Les itinéraires avec plus de dix événements de transmission dérivés sont affichés en rouge, les itinéraires avec plus de trois sont affichés en bleu et les itinéraires avec un à trois sont affichés en turquoise.
Résultats de l’étude
Les résultats de l’étude ont montré que les populations d’élevage de porcs ont été multipliées par plus de 200 au cours des 200 dernières années, l’augmentation maximale se produisant dans la seconde moitié des années 1920.Ème Siècle. Ces augmentations ont conduit à l’émergence progressive mais inquiétante de plus de 10 lignées au cycle biologique hautement pathogène. La forte densité de porcs d’élevage, généralement traités aux antibiotiques, a donné naissance à des lignées différentes et résistantes aux antibiotiques. S. suis présentent d’importants défis en matière de contrôle.
Des analyses d’échantillons provenant d’Espagne montrent que les porcs d’élevage et les sangliers sont des hôtes de souches de S. suis génétiquement très diverses, ce qui suggère que le lien entre le microbe et son hôte est ancien. Cependant, les analyses de datation génétique ont révélé que les six agents pathogènes les plus courants sont présents. S. suis Les tribus sont apparues au 19èmeÈme et 20Ème Des siècles, ce qui coïncide avec l’augmentation sans précédent de l’élevage d’hôtes.
“La conclusion selon laquelle ces données reflètent un changement écologique vers la pathogénicité dans au moins certaines de ces lignées est étayée par la preuve qu’elles ont coïncidé avec l’acquisition d’un îlot génomique associé à la pathogénicité (île 3).” Ceci est en outre étayé par les modèles de génome. réduction soutenue dans chacune des lignées pathogènes. Les comparaisons de différentes espèces bactériennes ont montré que la pathogénicité bactérienne est largement associée à des génomes plus petits et à moins de gènes.
Les analyses des capacités métaboliques des lignées pathogènes ont révélé qu’au moins deux îlots génomiques identifiés avaient considérablement amélioré leurs capacités à se développer au sein de l’hôte, les six îlots présentant des activités métaboliques accrues par rapport à leurs homologues plus bénins. Dans d’autres études bactériennes, la croissance au sein de l’hôte et le taux métabolique ont été associés à une virulence accrue, suggérant une tendance S. suis Adaptation à un cycle biologique plus virulent avec un plus grand potentiel de retombées zoonotiques.
« Les lignées pathogènes pourraient être mieux à même d’exploiter des régions spécifiques des amygdales que les lignées commensales et vice versa, réduisant ainsi la compétition au sein de l’hôte. Cela pourrait conduire à une ségrégation de ces populations et à une réduction du flux génétique entre elles, ce qui pourrait conduire à une réduction du génome dans des lignées plus pathogènes, car il existe moins de possibilités d’acquisition de gènes provenant de lignées commensales plus diverses.
Enfin, les analyses ont montré que l’électricité S. suis Les souches se caractérisent par un taux de propagation élevé et sont capables d’infecter rapidement un élevage porcin entier si un ou plusieurs individus infectés sont ajoutés. Le transport massif de bétail pose donc un problème supplémentaire : des souches virulentes auparavant endémiques se transmettent à travers les pays, voire les continents, et sont capables d’infecter de nouvelles populations hôtes qui n’ont que peu ou pas de résistance innée à leur égard.
«Nos résultats fournissent un cadre pour comprendre la diversité génomique de S. suis et sa relation avec la pathogénicité. Cela devrait être très utile à la recherche sur S. suis et au développement de stratégies visant à contrôler le fardeau de cette maladie sur la production porcine et la santé humaine. Étant donné que notre collection ne couvre qu’un petit sous-ensemble de pays dans le monde où les porcs sont élevés, des échantillonnages supplémentaires dans un plus grand nombre de pays et des échantillonnages plus approfondis au sein des pays, en particulier ceux ayant des populations porcines importantes et en croissance, sont nécessaires pour enquêter sur la présence d’agents pathogènes supplémentaires. les lignées à étudier sont géographiquement limitées ou n’ont émergé que récemment.
[ad_2]
Source